CO Oxidation on Pd(111): A First-Principles-Based Kinetic Monte Carlo Study
Recent experiments by Nakai et al. (J. Chem. Phys. 2006, 124, 224712) show that CO oxidation on O-precovered Pd(111) surfaces exhibits remarkably different reactivity at different temperatures, which correlates with structural changes in the atomic O overlayer. While the p(2 × 2) ordered phase is inert, the (v3 × v3) and p(2 × 1) phases that form at 320 and 190 K, respectively, display different apparent activation energies and reaction orders with respect to O coverage. Using kinetic Monte Carlo (kMC) simulations with activation energies and prefactors determined via density functional theory, we modeled this catalytic system to understand the origin of the changes in reactivity.
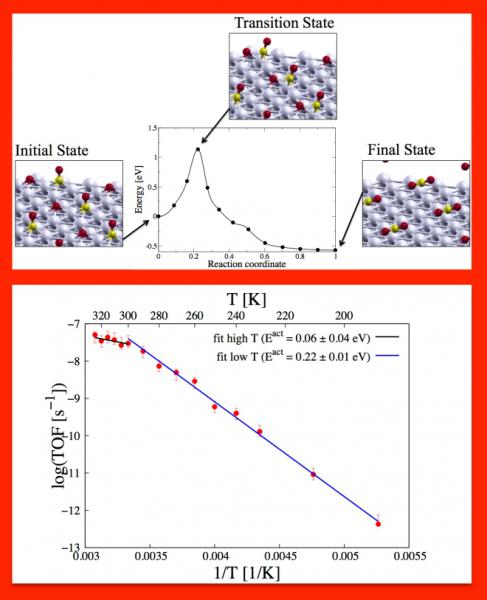
We explicitly account for lateral interactions among adsorbates and for their impact on the activation energies of the elementary processes. We have shown that a subtle interplay between the rates of CO adsorption/desorption and surface reaction is responsible for the changes in reactivity.At higher temperature, in particular, the order of the reaction with respect to O coverage is zero down to very low coverages. We find that beyond a certain coverage threshold, CO is adsorbed on sites where it experiences strong repulsion from the neighboring adsorbates, leading to a smaller adsorption energy and hence a significantly lower activation energy for CO oxidation. Ordering of the adsorbate layer, on the other hand, does not influence significantly the catalytic properties of the system.
S. Piccinin ; M. Stamatakis, "CO Oxidation on Pd(111): A First-Principles-Based Kinetic Monte Carlo Study", ACS Catal. in "ACS Catalysis", 4, 2143-2152 (2014) DOI
